Abstract
The preparation of lysergic acid 14C1-diethylamide is described. 14C1-Diethylamine was synthesised in three steps; sodium 14C1-acetate was treated with thionyl chloride giving 14C1-acetyl chloride which was reacted with ethylamine to give N-ethyl-14C1-acetamide. Reduction of this amide with lithium aluminium hydride gave 14C1-diethylamine which on treatment with a lysergic acid-imidazole complex gave the required product.
Introduction
As part of a study into the metabolism of lysergic acid diethylamide (LSD), the synthesis of carbon-14 labelled LSD has been investigated. Incorporation of carbon-14 the ergoline ring is not feasible and the two possibilities for labelling are the diethylamide side chain attached at C8 and the methyl group attached at N6. Carbon-14-LSD labelled in the diethylamide side chain has already been prepared1 and in metabolic studies2 only 4% of the carbon-14 was lost as 14C-carbon dioxide. Thus LSD was prepared containing carbon-14 in the diethylamide side chain.
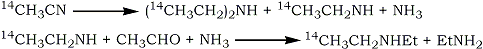
The method used by Stoll et al.1 for the synthesis of 14C1 -diethylamine involved catalytic hydrogenation of 14C-acetonitrile to give a mixture of diethylamine and ethylamine. The two amines were separated by counter-current distribution and the yield of 14C1-diethylamine was increased by reacting the ethylamine with ammonia and acetaldehyde.
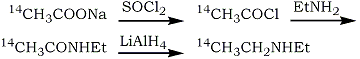
In view of the difficulties in purification and the necessity of synthesising the 14C-acetonitrile. The following scheme has been used in the present preparation:
No purification of the intermediates was necessary and the final solution of 14C1-diethylamine in ether was suitable for the subsequent formation of LSD. The incorporation of the 14C1-diethylamine into LSD was carried out by Stoll et al.1 by reaction with isolysergic acid azide. A more convenient method is that of Cerny and Semonsky3 which uses a lysergic acid-imidazole complex, formed in situ from lysergic acid and N,N'-carbonyl-diimidazole. This reaction is reported to give excellent yields when equimolar ratios of lysergic acid and amine are used.
Experimental
Chemicals
(+)-Lysergic acid was purified prior to use by solution in warm 2N ammonia followed by precipitation with 2N acetic acid4.
Chromatography
Thin layer chromatography was carried out on purchased aluminium roll, Silica gel F254 (E. Merck AG, Darmstadt, Germany). The Rf values for LSD, iso-LSD and lysergic acid in the solvent system, chloroform:methanol (3:2, v/v) are 0.74, 0.45 and 0.06 respectively. The spots were detected by fluorescence under UV light 254 nm) and by spraying with 1% N,N-dimethylaminobenzaldehyde in ethanol:HCl(aq) soln. (sp. gr. 1.18) (1:1, v/v).
Gas liquid chromatography was carried out on a Hewlett Packard Chromatograph and M. Scientific 402 High Efficiency Gas Chromatograph) fitted with a flame ionisation detector. A 5 ft glass column (3 mm I.D.) packed with PEG 6000 (10%), KOH (2%) on Chromosorb G (80-100 mesh) was used. Operating conditions: for N-ethylacetamide column temperature 150°C, N2 flow rate 150 ml/min, retention time [X] min; for diethylamine, column temperature 75°C, N2 flow rate 30 ml/min, retention time, 1.5 min.
Radiochemical Techniques
Radioactivity was determined with a Packard Tri-Carb liquid scintillation spectrometer (models 3214 and 3320) using a dioxane-based scintillator fluid@5. Scans of thin-layer chromatograms were made with a Packard radiochromatogram scanner model 7200). Also 0.5 cm wide sections of the absorbent were scraped into scintillation vials, scintillator fluid added and the radioactivity counted in the spectrometer.
Synthesis a) 14C1-Diethylamine
Thionyl chloride (160 mg) in dry ether (1 ml) was added to sodium 14C1-acetate (7.25 mg; 5 mCi) dried over P2O5 under vacuum. The mixture was stirred for 20 min at room temperature and then similarly dried sodium acetate (63 mg) was added. After stirring for a further 20 min the solvent was removed and the solid washed with dry ether (3x1 ml). The pooled solution was stirred, ethylamine (300 mg) in dry ether (2 ml) added slowly drop by drop and after cooling to 0°C the precipitate was filtered off. The filtrate was then evaporated at 0°C by a stream of dry nitrogen; the residue was taken up in dry ether (2 ml) and analysis showed a chemical yield of N-ethylacetamide of 55% (GLC) and a radiochemical yield of 48% (liquid scintillation counting, The solution of N-ethyl-14C1 -acetamide in dry ether (1.5 ml) was added drop by drop to a stirred suspension of LiAlH4 (160 mg) in dry ether (4 ml) at 20°C and the resulting mixture was stirred at room temperature for 16 h. The suspension was then boiled under reflux for 3 h and after cooling to 0°C, water was added drop by drop to decompose the excess LiAlH4. A violent reaction occurred with loss of some radioactivity from the top of the condenser. When the reaction stopped. the mixture was transferred to a stoppered test tube, washed in with 10% NaOH, and saturated with NaCl. The mixture was then shaken vigorously and the ether layer removed after it had separated. The aqueous phase was re-extracted with ether (4x2 ml) and the pooled extracts dried over Na2SO4. Analysis gave a chemical yield of diethylamine of 31% (GLC) and a radiochemical yield of 32% (liquid scintillation counting) with respect to sodium acetate.
Synthesis b - Lysergic acid 14C1-diethylamide
(+)-Lysergic acid (98 mg) dried over P2O5 under vacuum was suspended in dry DMF (8 ml) and N,N'-carbonyldiimidazole (252 mg) was added. After stirring the solution for 30 min in the dark at room temperature. [14C1]-diethylamine (19 mg, 1.6 mCi) in dry ether (9 ml) was added drop by drop. The extent of reaction was determined (TLC) and a further quantity of diethylamine (7 mg) was added to increase the yield of LSD. After a further 2.5h stirring, the solvent was removed by evaporation under reduced pressure, and the residue dissolved in 2% (+)-tartaric acid (30 ml). This acidic solution (pH 4) was extracted with ether:ethanol (9:1, v/v) (4x25 ml). the pH adjusted to 9 with 10% NaOH and re-extracted with ether:ethanol (9:1, v/v) (6 x [X] ml). TLC showed that almost all the LSD and iso-LSD was present in the basic extract which was then evaporated to dryness under reduced pressure, taken up in the minimum amount of methanol and streaked onto 4 20x20 cm Silica Gel H plates (0.75 mm). pre-eluted twice with methanol. After elution with chloroform:methanol (1:2, v/v) the two fluorescent bands were scraped off. eluted with methanol and assayed by TLC and radiochromatogram scanning. This showed traces of iso-LSD in the LSD and also some LSD in the iso-LSD.
The 14C-LSD was purified by crystallisation from diisopropyl ether after exhaustive drying over P2O5 under vacuum. The yield was 29 mg (26%). specific activity 13.59 µCi/mg). A second crop of 14C-LSD of lower specific activity was obtained by the addition of LSD ( 25 mg) to the mother liquor of the above preparation, followed by recrystallisation (yield 25 mg, specific activity 2.78 µCi/mg).
Purity of 14C-LSD
The mass spectrum (Varian MAT CH5) and IR spectrum (Perkin Elmer Infracord 37) are identical with a reference compound (Sandoz Ltd., Basle, Switzerland) and thin-layer chromatography in five systems [A, chloroform:methanol (3:2, v/v); B, acetone:chloroform (4:1, v/v); C, methanol:dioxane (3:2, v/v); D, chloroform: methanol:acetic acid (60:40:1, v/v); E, acetone:chloroform:NH3(aq) soln. (sp. gr. 0.88) (50:40:1.5, v/v)] gave only one spot. Radiochromatogram scanning and counting sections in 3 different solvent systems (A, B and C) gave a radiochemical purity of ~97%.
Discussion
This method of lysergic acid 14C1-diethylamide synthesis has advantages over the method of Stoll et al.1The two counter-current distribution procedures and the column chromatographic purification of LSD have been eliminated with a considerable saving in time. The preparation is also carried out on a much smaller scale, enabling a higher specific activity in the final product.
The yield of N-ethylacetamide can be increased if acetic acid is used as starting material. In this case, acetyl chloride is obtained in a similar yield (~90%) but the reaction with ethylamine proceeds more smoothly to give a higher yield of N-ethylacetamide (85% as compared with 55-65%)6. The yield on the reduction stage is poor (~60%) but this is probably due to losses during destruction of the excess lithium aluminium hydride, accentuated by the small scale of the preparation. Again the yield on the final stage, preparation of lysergic acid 14C1-diethylamide, is low. This is due to formation of iso-LSD and losses in chromatography and crystallisation.