Many forensic scientists who are avid viewers of the TV series "Quincy" cannot help cringing each time the protagonist peeks through a microscope and, without a moment's hesitation: "Eureka!" We sink a little lower in our seats as he rips a gas chromatogram from a recorder. studies it intensely for several nanoseconds, then slaps his right temple with the palm of his hand: "Of course! I should have known it from these yellow spots on his big toe!"
The speed and bull's-eye accuracy displayed by Dr. Quincy each week. although intended to keep the show moving at a pace suited to the average TV audience, unfortunately reflects and may even reinforce—the image that much of the community has of forensic scientists: that with little more than a microscope and a reagent that produces specific colors with every compound imaginable, the forensic scientist can produce a positive identification in just a few minutes.
Although we in the real world are frequently exposed to pressures not unlike those depicted on "Quincy." we have learned only too well that painstaking and time-consuming effort is often required to solve a tough analytical problem. Even after we piece together a molecular structure that appears to have analytical properties consistent with our data, we must subject it to what is oftentimes a laborious process of verification.
We at this laboratory are often asked how we go about identifying an unknown. In our college days, unknowns generally consisted of several grams of a single substance, identifiable by use of a variety of classical procedures, and for which an abundance of reference data was readily accessible. Forensic drug exhibits, however, usually consist of powders, liquids, and amorphous masses that are seldom obtained front reagent bottles. but, more likely, from scrapings of glassware in illicit laboratories or from matchbooks that have chanced hands under cloak-and-dagger circumstances.
We consider ourselves fortunate if we have gram quantities to tack with. and extremely fortunate if we discover that we have but a single ingredient to consider.
Since the majority of unknown exhibits that we examine consist of illicitly manufactured drugs or reaction intermediates, we can be assured of much work in determining which ingredients of forensic significance may be present, either as finished products or as substances that may be reacted to form them.
The initial approach to the identification of the sample components depends primarily upon the following factors: (1) the quantity of material submitted; (2) its known history; and (3) the preferred analytical procedures, instrumental and otherwise, of those assigned to the case.
We have to maintain complete flexbility in our approach as we gather information on the nature of the exhibit. Where a sizable amount of sample is available. for example, attempts are generally made to analyze separate portions by microscopy; thin layer chromatography; and mass, magnetic resonance, and infrared spectrometries. In many instances, the compounds of forensic interest are identified within a matter of hours, sometimes minutes, by a combination of interpretation and comparison of data with those obtained from reference substances.
The "China White" Case
In the matter of "China White" we were confronted with an impure, complex material with which the forensic community had no prior experience. It was available, initially, in only minute concentrations, and its identification was of the greatest urgency. Among drug users word had begun to spread of a synthetic substitute for heroin. Among law enforcement people a macabre aspect became prominent; people were dying and no one knew why.
Our first encounter with China White occurred last fall with a sample arriving from California. Although it had been implicated in a death where symptoms of analgesic overdose were exhibited, no drug could he detected either in it or in the victim's body fluids. Our initial examination of the powder revealed lactose and nothing more. Subsequent analysis by GC/MS of a highly concentrated extract. produced a weak spectrum that was totally unfamiliar and, unfortunately, whatever produced this spectrum was present only at very low concentration levels.
A short time later, however, an exhibit was referred to us that consisted of about 200 mg of powder having a greater concentration of this same substance. It was accompanied by a mass spectrum from GC/MS analysis of a methanolic extract of the exhibit and by an infrared spectrum of a hydrochloride salt prepared from the suspected active principal.
The mass spectrum resembled that obtained from the earlier exhibit. The infrared spectrum bore similarities to that of amphetamine; however, a band attributed to carbonyl was present, its location suggesting a tertiary amide (Figure 1).
The illicit laboratory from which the powder was thought to originate was believed to he engaged in the manufacture of methamphetamine. Indeed, a toxicological study conducted on an apparent drug overdose victim associated with that laboratory failed to reveal anything but amphetamine or methamphetamine. However, the quantities of these drugs were well below overdose levels.
Upon receiving the powder, we dry-extracted a substantial portion with deuterochloroform, then filtered it to remove the lactose. Examination by 1H-NMR revealed the presence of at least three ethyl groups, a possible cyclic group, and aromatic protons nearly equal in number to all the others (Figure 2). Following extractions from the chloroform with deuterated water and backwashes with additional chloroform, it was clear that we were dealing with three major components, one of which was a small compound with a partitioning preference for water. It contained an ethyl group, but gave no indication of aromaticity. The other compounds, however, exhibited a distinct preference for chloroform, although not in the same degree, and each contained both ethyl and aromatic groups. In addition, citrate was observed, present either as a mixture of citric acid and an inorganic salt, or in loose association with one of the major components.
It was evident from the weakness of the 1H-NMR spectrum that, although our exhibit had a greater concentration of suspected active principal than the one received originally, we had a total of no more than about 1 mg of this compound to work with. The limitations this would place on our analytical flexibility would undoubtedly delay the desired outcome.
Another portion of sample was subjected to GC/MS. Two major chromatographic peaks were observed. with several minor ones appearing between them. The early major eluant produced a fairly uncomplicated spectrum (Figure 3). With a base peak at m/z 93, an apparent molecular weight of 149, a small but significant fragment at. m/z 120, and fragmentation characteristic of a phenyl group (m/z 77, 65, 51, 39), very little was required to complete the picture. The other major component, however, produced the mass spectrum that had stymied us previously (Figure 4). We felt. certain that this was the one to tackle. Assuming a molecular weight of 259 from the fragment of highest observable m/z, we spent some time in trying to find clues in the fragmentation that would allow us to assemble building blocks adding up to 259. At this point it was clear that we had to make a more meaningful determination of molecular weight. The technique most readily available to us for this purpose was that of chemical ionization mass spectrometry (CIMS). Attempts Would also be made to see if the major components could be separated by simple extraction procedures so that we might obtain more meaningful 1H-NMR spectra and also to see if we could learn more about the structural functionalities of the major components from partitioning characteristics.
GC/CIMS provided a major breakthrough for us: Although the early eluter was indicated to have a molecular weight of 149, the late eluter appeared to have one of 350. The loss of 91 mass units to produce the 259 fragment in the E.I. mass spectrum might tentatively be ascribed to loss of a benzyl group. The presence of an intense m/z 91 ion in the E.I. spectrum further supported this hypothesis. A proposal that the m/z 110 fragment resulted from a loss of 149 from the m/z 259 ion and an indicated relationship to the early eluting compound was especially attractive despite the fact that charged and neutral 149 fragments of great. structural variety are not. uncommon in the mass spectrometry of organic molecules.
The mass 149 compound was found to extract as a neutral. The mass 350 compound, already known to have an amine function from its observed tendency to form a hydrochloride salt, was, predictably, found to he extractable from alkaline solution and also as a hydrochloride ion pair from acid solution.
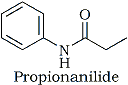
The remaining pieces of the mass 149 compound could now be determined with greater assurance. With an odd molecular weight of 149 there had to he at. least one nitrogen atom. We knew we had an ethyl group; a nitrogen had to he attached to the phenyl ring in order to produce an m/z 93 ion (assuming hydrogen transfer). Since the original 1H-NMR spectrum favored attachment of an aprotic carbon atom to the ethyl group and because of the presence of an m/z 57 ion, the existence of a carbonyl was all but assured. The pieces fit together nicely to form propionanilide. Further support for the structure was obtained from the 1H-NMR spectrum of the isolated compound1. With a reported LD50 of 1100 mg/kg,2 however, this compound could hardly qualify as a toxic substance, much less be the potent drug we were looking for.
In the 1H-NMR spectrum of the hydrochloride salt, prepared from the mass 350 compound (Figure 5), the presence of an anilido group was further supported. With most of the molecule tentatively accounted for, our attention was now focused on the m/z 110 fragment. We knew from the even molecular weight of the compound that, with only one nitrogen accounted for, there had to be another one here, or, with decreasing likelihood, three, five, or seven. Integration of the 1H-NMR spectrum told us that we probably had approximately 30 hydrogen atoms. Further consideration of the mass spectral and NMR data strongly suggested the presence of a piperidine ring, and a methyl group attached to CH, possibly at the 3-position of the piperidine ring.
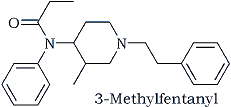
The pieces were now beginning to come together. The next step was to search Chemical Abstracts for information on highly toxic compounds that could conceivably fit the proposed structural data. Armed with empirical formulas derived from the tables of Beynon and Williams3 that were consistent with presumed molecular weight and proton count, the search commenced. After much diligence, a likely candidate emerged: 3-methylfentanyl.
This compound seemingly fit the bill from the standpoint of structural as well as toxicological characteristics. The piperidine structural moiety was sufficiently consistent with mass and 1H-NMR spectral data that it was felt that a tentative identification had been made. It appeared that our search had just about come to an end.
However, the following remained for us to do:
- Confirm the proposed structure with the aid of spectral comparison with a reference compound.
- develop methodology that could be employed by other forensic laboratories for rapid screening,
isolation, and identification of the active component; and - obtain all available information from the literature on the proposed compound.
Upon further study of the data, however, an inconsistency with the proposed structure became apparent. A weak 1H-NMR signal at about 4.7 ppm, representing a single proton believed to be in the 4-position of the piperidine nucleus, produced a pattern that was seemingly inconsistent with the assigned structure. In both salt and basic forms of the compound, it appeared as a "triplet of triplets," a result of two large coupling constants and two small ones. Since this could arise only from a pair of axial-axial (and/or geminal) proton interactions. and a pair of axial-equatorial proton interactions, this meant that substituents could not be present in both the 3- and 4-positions. The possibility of overlapping "doublets of triplets", so spaced to produce an apparent "triplet of triplets” from a mixture of cis and trans isomers (axial and equatorial 3-methyl, respectively), had to be discounted after serious consideration because of the theoretically vast differences in coupling.
By now we had some standards available for comparison and some informative reprints4-6. The 3-methylfentanyl standard, following purification by HPLC, did not compare with the unknown, although some structural similarities were evident. The 2-methylfentanyl, although it produced a 1H-NMR pattern for the 4-position proton similar to that of the unknown, was not the compound of interest. In view of the structural assignments for which we had solid support. where could we reassign this methyl group? Only two places remained: alpha and beta to the piperidine nitrogen. The beta position seemed highly unlikely because of the predicted effects on the mass spectral fragmentation: intense ions at m/z 105 (in addition to the m/z 91 ion) and at m/z 96 (at the expense of the m/z 110 ion), neither of which was evident. This left us with the alpha isomer, which we could find no reason to discount.
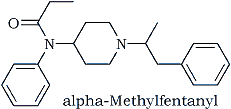
All efforts now were concentrated on obtaining a reference standard of this material as quickly as possible. Within days it was synthesized, purified, and subjected to spectrometric examination. It was examined as a base and as a hydrochloride salt. The outcome? A match with the sample in all respects!
What of the water-soluble component that contained an ethyl group and was not observed by GC-MS? It was identified as propionic acid. It was believed to have arisen either as a by-product of the synthesis or as a decomposition product.
As the case drew to a close we became subjected to a steady stream of reporters and photographers from the various media. The last of them, an established science reporter from s radio network, discussed the case with us at great length. Toward the end of the interview, however, I (T.C.K.) was abruptly disturbed by a reminder of the legacy of Dr. Quincy and his fictional predecessors. "Why," asked this otherwise seasoned reporter, "did it take nearly a month to come up with an answer?"