A novel isomerisation of epoxides to carbonyl compounds by methyl iodide and sodium iodide in dimethylformamide was described earlier1. A study of the analogous reaction in dimethyl sulphoxide has now revealed features of its mechanism, linking it with other reactions involving activated dimethyl sulphoxide2.
In a typical experiment an approximately 0.07M-solution of the epoxide in dimethyl sulphoxide containing n-propyl iodide (0.35M) and sodium iodide (0.35M) is kept at 80°C for 3 hr. The dark reaction mixture contains iodine (which is not catalytically active) but, after aqueous work-up, it affords the carbonyl compound(s) in high yield; thus octan-4-one (100%) is obtained from cis-4,5-epoxyoctane, octanal (80%) and octan-2-one (4%) from 1,2-epoxyoctane, phenylacetaldehyde (85%) and acetophenone (4%) from styrene oxide, and cyclohexanone (90%) from epoxycyclohexane.
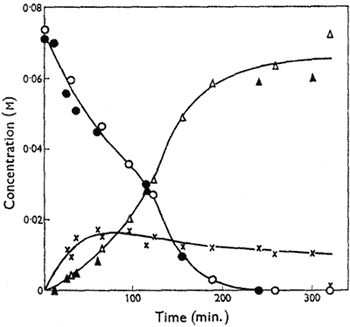
Figure.
Variation of the concentrations of cis-4,5-epoxyoctane
(circles), octan-4-one (triangles) and, by difference, the
metastable intermediate (crosses) with time in DMSO at
70°C. Open symbols signify catalysis by n-propyl iodide
(0.034M) and solid symbols n-propoxydimethylsulphonium
tetraphenylboron (0.033M). [NaI] = 0.34M in both cases.
The kinetics of isomerisation of cis-4,5-epoxyoctane at 70°C have been studied; 2% water was added to eliminate rate variations from imperfect dryness, but it has little effect on the yield of the reaction at this level. Reaction mixtures were partitioned between chloroform and water and the organic components analysed by gas chromatography using a 100 ft. stainless-steel capillary column with 18% dinonyl phthalate as stationary phase and diglyme as internal standard. The rate of disappearance of the epoxide is substantially faster than the rate of appearance of octan-4-one, indicating the formation of a metastable intermediate which is not detectable by these techniques. Over the first 50% of reaction, the rate of disappearance of the epoxide is proportional to the epoxide concentration (In the later stages of the disappearance of the epoxide the rate increases, an effect which appears to be related to the formation of appreciable amounts of octan-4-one), and it increases with increasing concentration of n-propyl iodide, though not proportionately; it is independent of the concentration of sodium iodide. However, sodium iodide has a marked accelerating effect on the production of octan-4-one, and, indeed, terminal yields of ketone fall significantly when no sodium iodide is added to the reaction mixture.
Alkyl iodides are known to alkylate dimethyl sulphoxide either on oxygen to form alkoxydimethylsulphonium iodides or on sulphur to form alkoxydimethylsulphonium iodides3,4. Trimethyloxosulphonium iodide5 is ineffective in isomerising epoxides under conditions where methyl iodide gives a smooth conversion. On the other hand, n-propoxydimethylsulphonium tetraphenylboron converts cis-4,5-epoxyoctane into octan-4-one by processes having an identical kinetic form and similar rate coefficients to those found for the isomerisation induced by the same concentration of n-propyl iodide (see Figure). It is well established that alkoxydimethylsulphonium ions are converted by iodide ion into alkyl iodide and dimethyl sulphoxide4,6, but we have established by NMR spectroscopy that, under our reaction conditions, some of the alkoxysulphonium salt remains.
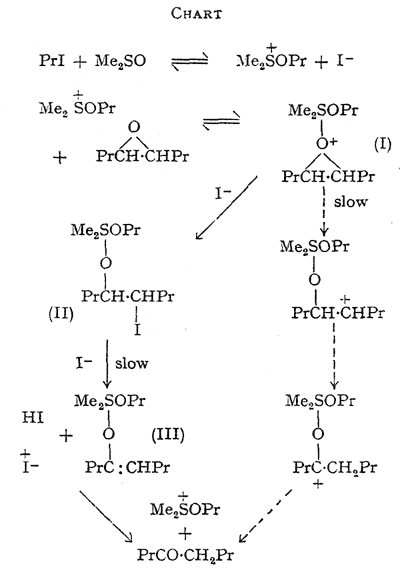
The foregoing data are accommodated in an hypothesis, the essential features of which are outlined in the Chart. The slow step in the main route for epoxide disappearance is seen as the decomposition of an oxonium complex (I) formed between the alkoxysulphonium salt and the epoxide giving the quadrivalent sulphur intermediate (II). ß-Elimination of hydrogen iodide from (II), initiated by iodide ion, would yield a second quadrivalent sulphur derivative (III) of the enol form of octan-4-one (Consistent with this, the epoxide equally labelled with tritium at C-4 and C-5 yielded octan-4-one with 57% retention of the label.
This indicates that the major isomerisation route involves intermolecular hydrogen transfer but that some intramolecular transfer, expected in a carbonium-ion mechanism, may take place, see Chart). Hydrolysis of (II) on work-up would yield an iodohydrin, which is not detectable by our analytical method. Significantly, this iodohydrin, prepared separately, yields octan-4-one when treated with sodium iodide in dimethyl sulphoxide at 70°C. The direction of isomerisation of the unsymmetrical epoxides 1,2-epoxyoctane and styrene oxide is explained, since the major products are analogous to those expected from acid-catalysed opening of the three-membered ring.
According to this hypothesis, other Lewis acids capable of co-ordinating with the epoxide should promote the isomerisation of cis-4,5-epoxyoctane in dimethyl sulphoxide containing iodide ions. Hydrogen iodide itself is effective, but both rate and yield are lower than with n-propyl iodide. On the other hand, chlorodimethylsulphonium tetraphenylboron (New compound, mp 108°C, prepared from phosgene and dimethyl sulphoxide with subsequent addition of sodium tetraphenylboron in aqueous methanol at 0°C.) is even more effective than n-propyl iodide, and the reaction mixtures are colourless. The tetraphenylboron salt prepared likewise from dimethylformamide (Cl-CH=N+Me2B-Ph4) is also a very effective catalyst, and this supports our initial suggestion that the reactions in dimethylformamide and dimethyl sulphoxide are essentially similar. Alkyl iodides and sodium iodide do not significantly catalyse the isomerisation in sulpholane, acetonitrile, or butan-2-one.