Abstract
Chromatographic purification is an integrated part of organic synthesis. The Dry Column Vacuum Chromatography presented here, has excellent resolving power, is easily applied to large scale chromatography (up to 100 g) and is fast. Furthermore, the technique is economical and environmentally friendly due to significant reductions in solvent and the amount of silica used. Therefore, it is an excellent alternative to the commonly used Flash Column Chromatography for purification in organic synthesis.
In our efforts to synthesise large quantities of the nucleoside analogue LNA,1,2 we regularly purify quantities of up to 100 g of highly functionalised nucleoside and carbohydrate derivatives. Conventional silica gel column chromatography techniques including Flash Column Chromatography3 afford poor separation of amounts exceeding 10 g demanding purification of the reaction mixtures after division into several minor batches. This is unfeasible due to the large quantities of silica gel, solvents and time required.
In our search for a simple and efficient method for large scale separation of complex mixtures, we re-discovered and further developed a technique first described by L. M. Harwood called “Dry Column” Flash Chromatography.4 This technique has mainly been used for small to medium scale purifications (< 2 g) with separation power rivalling that of analytical TLC. We have now adapted this technique to large scale chromatography (over 10 g compound mixture) without losing the excellent resolving power observed with smaller amounts on smaller columns. We have prepared and eluted large columns (diameter 10–13 cm) with up to 100 g of complex mixtures in 1–2 h depending on the number and size of the fractions collected, the efficiency of the pump and the kind of silica type used. We have chosen to name this improved version of Harwood’s technique Dry Column Vacuum Chromatography (DCVC) since “Flash” is misleading in this context.5
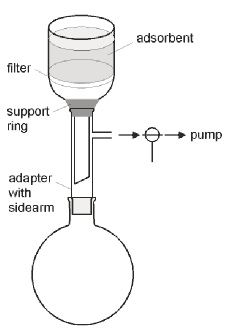
Figure 1
Original setup
suggested by L. M. Harwood
The general experimental setup suggested by the inventor4,6 and others7,8 consists of glassware found in any organic chemistry laboratory. However, the setup is not practical as it is necessary to disassemble it for each fraction (Figure 1), which is tedious and time consuming.
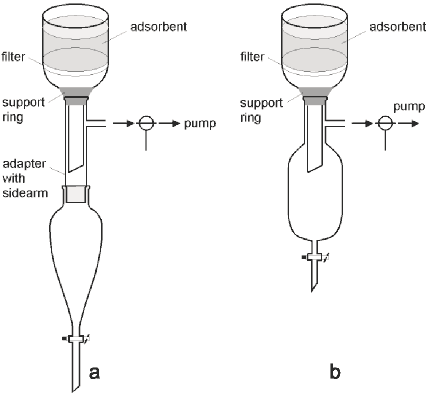
Figure 2
a Setup with standard glassware;
b Setup with specialised glassware
The setup is significantly improved by using an ordinary separatory funnel,9 an adapter with a sidearm and a three-way stopcock attached on the sidearm allowing one to let air into the funnel without turning the pump off (Figure 2a). The only drawback with this setup is that with large sintered glass funnels (diameter over 10 cm) it tends to get very high and as a consequence impractical. We have solved this problem by designing a compact vacuum proof receiver (Figure 2b). Our adaption of the experimental setup has significantly reduced the time required for DCVC and provides a construction that is easy to handle for large as well as small scale chromatography.
The experimental procedure can be summarised as follows (see experimental section for detailed procedure):
- A sintered glass funnel of the appropriate size10 is filled with approximately 6–7 cm of loose silica and tapped to give a level surface.
- Vacuum is applied and the surface is pressed firmly to give a completely level, well compacted bed approximately 4.5–5.5 cm high.
- The column is checked for voids and channels by pouring n-heptane onto the silica bed while applying vacuum. If the column is packed properly, the solvent will descend in a horizontal line.
- The mixture to be separated is dissolved in an appropriate low boiling solvent (eg. ethyl acetate, methanol) and pre-adsorbed on silica.11 The silica is added as a thin uniform layer on top of the column and step (2) is repeated.
- The column is gradient eluted with a suitable solvent system and fractions are monitored by TLC.
Several problems can arise due to evaporation of solvents during chromatography. The most significant being difficulty in performing a controlled gradient elution and water condensing on the glassware. This can be prevented by avoiding volatile solvents such as diethyl ether, dichloromethane and pentane. Mixtures of n-heptane, ethyl acetate, and methanol are recommended as they are easy to handle and remove after separation has been accomplished.
Elution of compounds is usually accompanied by frothing on the underside of the sinter. As a rule of thumb, a compound will elute in the solvent mixture where the Rf value is ca. 0.5 on analytical TLC.
When chromatographing a mixture of compounds (approx. 20–50 g) with a ΔRf ~0.05 (by TLC) on a sintered glass funnel with a diameter of 10 cm, we usually collect 100 mL fractions by gradient elution. Normally this yields compounds that are >95% pure by HPLC. If a better separation is desired a different solvent gradient and collection of smaller fractions usually improve the result.
In order to obtain good separation, the choice of the correct silica type is essential. Resolution is determined by particle size. As a rough guide, reduced particle size gives better resolution. On the other hand, too small particles will result in an extremely compact column that elutes very slowly. According to the inventor6 and others,7 TLC silica without the gypsum binder (5–25 µm) should be used for DCVC because it is cheaper than the silica used for regular chromatography (e.g. Flash Column Chromatography) and because “regular” silica is too free flowing to allow easy column packing. This is also true in our hands for Merck Silica Gel 60 (40–63 µm) used for Flash Column Chromatography because the normal packing procedure does not yield a sufficiently compact column. As a result, the solvent just pours through the columns affording very poor separation. If the column is packed using a high vacuum pump, compact columns with good separation can be prepared. However, this approach is undesirable for several reasons, most of all because of the long time required to prepare such a column.
If Merck Silica Gel 60 (15–40 µm) is used, the problems are avoided. We have experienced no problems when using Merck Silica Gel 60 (15–40 µm) instead of TLC silica (5–25 µm) and in our hands the same time is needed to prepare a column with either type of silica. Although it is difficult to reproduce the exact chromatography conditions, the data suggest that TLC silica (5–25 µm) without the gypsum binder offers a slightly better separation compared to Merck Silica Gel 60 (15–40 µm) as expected due to smaller particle size. TLC silica (5–25 µm) is about 10–15% cheaper than Merck Silica Gel 60 (15–40 µm). Furthermore, a column packed with TLC silica (5–25 µm) requires approximately 10–15% less silica by mass compared to Merck 60 (15–40 µm). However, a column packed with TLC silica becomes very compact and therefore significantly slower in eluting.
Our preferred silica is Merck Silica Gel 60 (15–40 µm) as it offers excellent resolution and gives considerably faster eluting columns than those packed with TLC silica.
A typical example of a compound mixture that was separated successfully is shown in Figure 3. An analytical TLC of the compound mixture in 10% n-heptane, 90% ethyl acetate (v/v) showed 4 compounds with Rf values: 0.45 (1), 0.25 (2), 0.20 (3) and 0.10 (4). The compounds were successfully separated and isolated on a 10 g scale in 1.5 hours including packing of the column, elution and evaporation of the solvent fractions containing the pure compounds. The column (diameter 10 cm, height 5 cm, Merck silica gel 60, 15–40 µm) was eluted with 0–100% ethyl acetate in n-heptane (v/v) with 5% increments in ethyl acetate concentration for each fraction collected (twenty-one 100 mL fractions). After isolation, the identity of the compounds depicted below was established by NMR. The desired compound 2 was isolated in 98+% purity by HPLC and 1H-NMR.
In conclusion, DCVC provides a robust, fast and economical method for large as well as small scale separations. In addition, the technique is superior in resolution power to Flash Column Chromatography because of the reduced diffusion during chromatographic separation. This means that each compound elutes in fewer fractions and that less cross contamination of fractions is observed.
We have used Dry Column Vacuum Chromatography for purification of a wide range of complex mixtures comprising highly functionalised nucleosides and carbohydrates with excellent results. It is possible to perform DCVC using mixtures of nonchlorinated, nonvolatile solvents such as n-heptane, ethyl acetate and methanol. This and the greatly reduced amount of silica used compared to other techniques reduces the enviromental impact of the process. Separation of compounds with ΔRf ~0.05 (by analytical TLC) should at least be expected, but with experience separations as efficient as those with analytical TLC can be achieved.
Chromatography equipment was assembled as depicted in Figure 2a or 2b. Analytical TLC was performed using Merck 5554 silica 60 aluminum sheets. The silica used for DCVC was Merck 15111 silica gel 60 (15–40 µm) and Merck 9385 silica gel 60 (40-63 µm) purchased from Merck Eurolab. TLC silica without binder (5-25 µm) was purchased from Sigma-Aldrich Denmark. Solvents were HPLC grade from LABSCAN. Vacuum was applied with a Vacuubrand MZ 2C diaphragm vacuum pump (1.7 m3/h, 10 mbar) instead of a water aspirator for enviromental reasons. Sintered glass funnels (porosity 3) with diameters up to 17 cm are commercially available.
Dry Column Vacuum Chromatography
Column Packing: As a rough guide, 1 cm2 of silica surface can be charged with up to approx. 500 mg of compound mixture. However, the sample size per cm2 can be increased substantially if less resolution is required. The glass funnel should be at least 7 cm from the sinter to the lip allowing each solvent fraction to be added in one batch instead of slowly being poured onto the surface of the silica. A cylindrical sintered glass funnel (porosity 3) is filled with 6–7 cm of loose Merck 15111 silica gel 60 (15–40 µm) and then tapped to give a level surface of silica. The glass funnel is placed in a Buchner flask and vacuum is applied. The silica is pressed firmly with a flat object (eg. glass or rubber stopper) to give a flat, well compacted silica column approx. 4.5-5.5 cm high. Special care should be taken to compress the silica at the circumference of the glass funnel with a spatula. The column is checked for voids and channels by pouring n-heptane onto the surface (protected by filter paper) while applying vacuum. If the column has been properly prepared, the solvent will descend in a horizontal line. If this is not the case, the column must be sucked dry and the above packing procedure repeated. With acid sensitive compounds, the silica can be pre-treated with a mixture of 1-5% Et3N, n-heptane (v/v) during the packing procedure.
In our experience, columns higher than approx. 4.5-5.5 cm do not improve resolution and shorter columns should be avoided because of significantly reduced separation power. Short columns (app. 1-3 cm high) can be quite good for very simple separations but truly provides more of a filtration than an actual chromatography step.
Sample Application: While preparing the column, the compound mixture should be pre-adsorbed on Merck silica gel 60 (15–40 µm).11 The mixture is dissolved in a small volume using an appropriate solvent (e.g. mixtures of EtOAc and MeOH) and the silica is added. To obtain optimal separation it is desirable to apply the sample on the column in as thin a layer as possible. For this reason, no more than ca. 1:1 (w/w) silica should be used for pre-adsorbtion. The solvent is removed from the slurry under reduced pressure on a rotary evaporator. A splashguard should be used since silica has a tendency to bump when it is almost dry. The slurry should not be evaporated to complete dryness in a single step since this may result in a hard crust. Instead the silica should be scraped down the sides of the flask before the last solvent has been removed (if a coarse type of silica is used for pre-adsorption it is most easily removed from the flask). When the silica is dry, it is transferred to a mortar and ground to give a fine powder that is added in a thin uniform layer on top of the column. Vacuum is applied and the surface is pressed firmly as in the column packing step.
Solvent System: The column is developed by gradient elution using a suitable solvent system. For most separations, mixtures of n-heptane, EtOAc and MeOH are excellent. The least polar solvent mixture is added first followed by solvent fractions typically with 1-10% increments in the most polar component. Our preferred solvent system for most separations is 0-100% EtOAc in n-heptane (v/v) - with increments of 5%. For very polar mixtures, it is preferable to start in a more polar solvent mixture of n-heptane, EtOAc and shift to solvent mixtures of EtOAc, MeOH when 100% EtOAc is reached.
Elution: When the solvent system has been decided, vacuum is applied to the column. The first solvent fraction is poured on the filter paper protected surface of the column. While the fraction is eluting the next fraction is prepared. The solvent fractions should not be prepared beforehand as this is too time consuming and evaporation can result in poor gradient elution. The first solvent fractions are usually lost due to adsorption on the silica. When the fraction has been sucked through the column (i.e. the solvent is dripping slowly from the column), air is allowed into the setup by turning the three-way stopcock (Figure 2) and the solvent fraction is collected. Vacuum is applied again and the next solvent fraction is poured onto the surface of the column. Elution of compounds is monitored by TLC.
As a rule of thumb, compounds will elute in the solvent mixture where they have an Rf value of approximately 0.5 on analytical TLC. Elution of compounds often co-occurs with frothing on the underside of the sinter. The size of the fractions and the increase of the more polar component in the solvent system varies with the desired separation. Smaller fractions and a slow increase in polarity improves the resolution significantly but also takes considerably longer time.