Abstract
High yields in the methylenation of catechols by methylene chloride are obtained by song a polar aprotic solvent for reaction and maintaining low concentrations of the catechol dianion.
When Fittig and Remsen1 first methylenated a catechol (3,4-dihydroxybenzoic acid), by heating it with methylene iodide and potassium hydroxide, they remarked 'Die Ausbeute war eine sehr geringe'. Similar comments tended ever since to punctuate descriptions of methylenation of catechol and its derivatives, and uncontroverted claims to satisfactory yields have been few, though many attempts at improvement have been made.
Benzo-1,3-dioxole [methylenedioxybenzene, (I)] was first made from catechol, methylene iodide, and ethanolic sodium ethoxide in 18962, and numerous preparations have since been described. The ionizing base for catechols has always been an alkali hydroxide, alkoxide or carbonate; the methylenating agents have variously been methylene iodide in alcohol2-4 or acetone5, methylene bromide4, 'methylene sulphate'6,7, methylene chlorobromide8 and methylene chloride4,9-12. The best yield (69%) of benzo-1,3-dioxole was obtained with methylene iodide, methanolic potassium hydroxide, catechol and copper bronze, heated and shaken together at 100-110°C for 18 hr. in a sealed tube. The yield from methylene chloride with these conditions was 53%.4
Methylene chloride is, of course, the halide of choice for large-scale preparation, and Laskina12 worked out a procedure for methylenating catechol with this halide at atmospheric pressure. With ethylene glycol as solvent, sodium carbonate as base, and with a temperature of 122-124°C, the yield was 38-43% after 8-12 hr. To maintain the high temperature, methylene chloride was introduced gradually.
The nature of the side-reactions that lower the yields of benzo-1,3-dioxole have been little explored. Gensler and Samour8 reported formation of the 'dimer' (II), and of two high-boiling oils (which had hydrogen contents suggesting to us that the solvent ethanol participated in their formation). An intractable tarry material generally accounts for most of the catechol not appearing as benzo-1,3-dioxole.
The formation of benzo-1,3-dioxole probably proceeds in two steps:
- A bimolecular nucleophilic displacement of halide ion from the methylene halide by catechoxide dianion (when the ionizing base is strong enough to form the dianion); yielding the haloguaiacol anion (III).
- An intramolecular displacement of a second halide ion from (III).
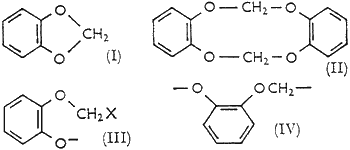
Methylene halides are not highly reactive to nucleophiles, which accounts for the slowness of the reaction as usually carried out. Unless halomethylation on carbon is a competing reaction, the anion (III) is a likely intermediate for all products eventually formed. The 'dimer' (II), for example, could arise by reaction of (III) with (a) itself, (b) catechoxide dianion and then methylene halide, or (c) methylene halide and then catechoxide dianion. Of these possible courses (b) seems most likely and (c) least likely, since catechoxide dianion is probably the most reactive nucleophile present and α-alkoxyhalides are much more reactive than methylene halides to nucleophiles. Successful competition of these bimolecular reactions with the intramolecular cyclization leading to (I) may seem unusual, but examination of models of the anion (III) indicates that the cyclization is stereochemically less favoured than, e.g. the analogous cyclization of a 4-halogenated alkoxide to a tetrahydrofuran. With normal bond-angles, the minimum distance of the anionic oxygen from the halogenated carbon is ca. 0.4 A greater in (III) than in a 4-halogenated alkoxide. The isolation of the 'dimer' (II) as a reaction product is a clear indication that bimolecular processes do compete with the cyclization; and because closure of the 10-membered ring in (II) is unlikely to be favoured, the operation of processes (a), (b) and (c) should lead to larger amounts of linear polymers in which the repeating unit is (IV). If, therefore, competition from bimolecular processes for the intermediate (III) is important, the formation of benzo-1,3-dioxole should be preferred at lower concentrations of catechoxide dianion. The concentration of (III) is thereby also lowered and the processes (a) and (b) become less favourable relative to the intramolecular cyclization. If process (c) is, as expected, unimportant, it would be less effective to lower the concentration of methylene halide.
However, the rate-limiting step in the overall reaction is undoubtedly the formation of intermediate (III), and this bimolecular step would also be slowed by reducing the concentration of catechoxide dianion. In the usual, rather concentrated, alcoholic media the reaction is already inconveniently slow, especially with methylene chloride; it would become impracticably so at higher dilutions. One must, therefore, use a solvent which greatly accelerates nucleophilic displacements by anions: this will have the effect of speeding up all stages of the process. Dimethyl sulphoxide and dimethylformamide are familiar examples of such solvents; because of its greater stability to alkali, dimethyl sulphoxide appears preferable.
Experiment provided strong support for the reasoning set out above. Catechol (initial concentration 2.5M) in dimethyl sulphoxide was heated with slight excesses of sodium hydroxide and methylene chloride in a bath at 120°C. The vigorous reaction was over in 10 min. and the yield of benzo-1,3-dioxole was 46%. At initial catechol concentrations of 1M and 0.67M, the yields were respectively 68 and 73%; with the most dilute solution, 70 min. were allowed for reaction. When dimethylformamide was the solvent, an initial catechol concentration of 2M led to a 54% yield of benzo-1,3-dioxole.
Methylenation in dimethyl sulphoxide was applied to some alkylcatechols at initial concentrations of 0.67M, and yields of 70-80% were obtained in conformity with the results from catechol. Methylenation of the parent phenol was then studied further, and the yield of benzo-1,3-dioxole was raised to 91% by adding catechol and sodium hydroxide separately, simultaneously, and slowly to a stirred, heated mixture of dimethyl sulphoxide and methylene chloride. The 'dimer' (II) could be found in small quantity among the by-products. The procedure was also applied to the preparation of piperonal.
Experimental
Benzo-1,3-dioxole (I)
A mixture of methylene chloride (100 ml.) and dimethyl sulphoxide (500 ml.) was stirred and heated to 125-130°C under a wide-bore Liebig condenser. The air was displaced by nitrogen and then pelleted catechol (5.5g) and pelleted sodium hydroxide (4.15 g.) were added simultaneously. This was done without the admission of air by placing the pelleted reagents separately in two tubes (15-20 mm. diam.) which had been sealed through the bottom of an inverted conical flask fitted to the top of the condenser. The two tubes were fitted with stoppers and side-arms for admitting nitrogen, and were both closed at the bottom by one flat piece of polytetrafiuoroethylene attached to a spindle, which was parallel to the tubes and was mounted in a gland between them. Rotation of the spindle through 90° allowed the pellets to fall simultaneously and cleanly down the condenser into the reaction mixture. Another 90° rotation closed the tubes in readiness for the next charge, which was added 5 min. later. Totals of 110 g. of catechol and 83 g. of sodium hydroxide were thus added during 105 min. After a further 20 min. methylene chloride (20 ml.) and sodium hydroxide (3 g.) were added and the stirring was continued for yet a further 70 min. The condenser was then replaced by a Vigreux column and a constant-take-off distillation head. Water (50 ml.) was added and the benzodioxole-water azeotrope was distilled off at 98-100°C, more water being added slowly as distillation proceeded. Benzodioxole ceased to separate as a heavy oil from the distillate after ca. 600 ml. of distillate had been collected. The aqueous distillate was extracted with ether (3x50 ml) and the combined oil and extract was dried and distilled to yield benzo-1,3-dioxole (111g, 91%) as a colourless oil; nD21 1.5377, bp 60°C/9 mm., 173-175°C/760 mm. Distillation of the residual reaction mixture at reduced pressure gave unchanged dimethyl sulphoxide (490 ml).
Homologues of Benzo-1,3-dioxole
4-Methylcatechol (12.4g.) and methylene chloride (10 g.) were dissolved in dimethyl sulphoxide (150 ml.) under nitrogen and powdered sodium hydroxide (8.3 g.) was added. The mixture was heated for 2 hr. under reflux in a bath at 120°C. Steam distillation then afforded 5-methyl-benzo-1,3-dioxole (9.5g, 70%), bp 196-197°C/753.5 mm., nD20 1.5308.
From 3-isopropylcatechol (15.2 g.) under identical conditions there was obtained 4-isopropylbenzo-1,3-dioxole (13.1g., 80%), bp 98°C/14 mm., nD20 1.5181 as a colourless oil.
Piperonal
The procedure used to make benzo-1,3-dioxole was followed, on one-tenth the scale. From 3,4-dihydroxybenzaldehyde (13.8g), piperonal (9.15 g., 61%) was obtained by steam distillationand identified by comparison with an authentic sample.
2,7-Dibenzo[1,3,6,8]tetroxecin (II)
From the non-volatile residues of benzo-1,3-dioxole preparations, a crystalline substance was isolated by extraction with alcohol and sublimation under reduced pressure. This substance, after recrystallization from acetic acid, had mp 261-262°C.
Comparison with a specimen kindly provided by Professor W. J. Gensler of Boston University confirmed the identity of this substance with 'methylenedioxybenzene dimer' (II) already reported.8