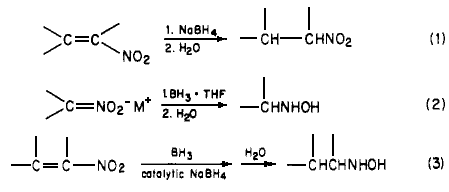
In connection with our ongoing program directed toward the synthesis of radiolabeled amphetamine derivatives1, we required a convenient method for the preparation of hydroxylamines, A survey of the literature indicated that the most convenient syntheses of N-substituted hydroxylamines involved the reduction of conjugated nitroalkenes2,3. Other methods include the reduction of oximes4 and nitro salts5 or the oxidation of amines6. The latter methods are involved and are not readily amenable to the synthesis of the desired target molecules.
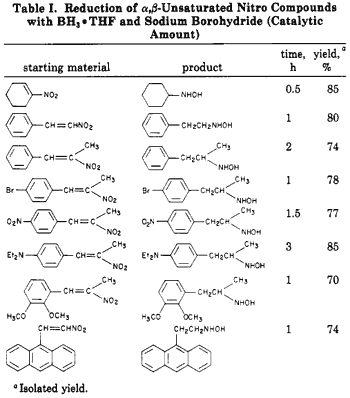
Since catalytic hydrogenation of conjugated nitroalkenes is reported to be a complex reaction2, we investigated the reduction of conjugated nitroalkenes utilizing lithium aluminum hydride3. Unfortunately, in our hands, mixtures were obtained under a variety of conditions. We then investigated the reduction of conjugated nitroalkenes with boron hydrides. It had been reported that the sodium borohyride reduction of a,ß-unsaturated nitroalkenes produces the corresponding nitroalkanes (Eq 1)7, In a later study, Feuer reported5 that nitro salts (nitronates) are readily reduced to hydroxylamines by borane complexes, Eq 2 (i.e, the nitro compounds are unreactive). These reactions presumably occur through a common intermediate 1, which can then be hydrolyzed directly to nitroalkane or reduced with a borane complex to yield hydroxylamine after hydrolysis, It occurred to us that these reactions could be utilized to prepare the desired hydroxylamine directly from the conjugated nitroalkenes.
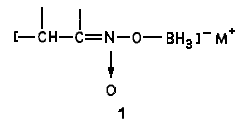
We have found that sodium borohydride catalyzes the reaction of borane complexes with a,ß-unsaturated nitro compounds, The reaction is straightforward. A catalytic amount of sodium borohydride is added to a normally unreactive mixture of the a,ß-unsaturated nitro compound and the borane complex at room temperature. The pure hydroxylamines are readily isolated in high yields (Eq 3). Our results are summarized in Table I.
Experimental
Table II.
1H and 13C NMR Data for Nitroalkenes.
All glassware was thoroughly dried in an oven and cooled under dry nitrogen just before using. THF was dried and distilled over CaH2 and LiAlH4 and kept under dry nitrogen. The BH3·THF solution was prepared and standardized according to the published procedure8.
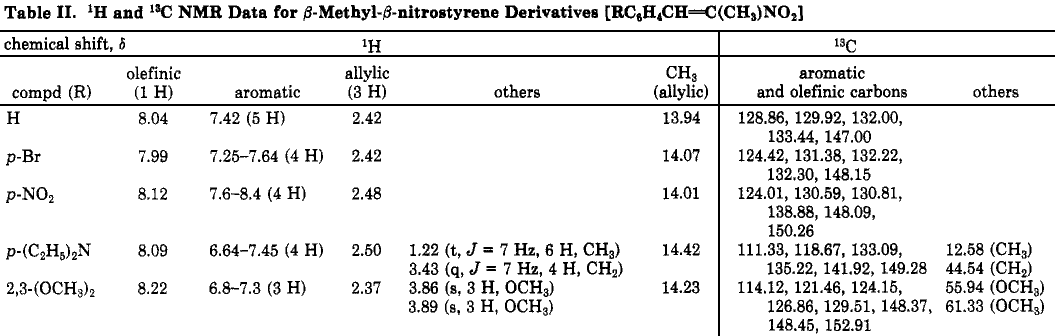
Commercially available samples (Aldrich) of 1-nitro-1-cyclohexene, ß-nitrostyrene, and 9-(ω-nitrovinyl)anthracene were used as received. Other nitro compounds were prepared via published procedures,9-11 and the spectral data are summarized in Table II.
![]()
Synthesis of N-Substituted Hydroxylamines
General Procedure.
The synthesis of N-hydroxy-2-phenylethanamine is representative
of the procedure employed.
A flame-dried, nitrogen-flushed, 250-mL flask, equipped with a septum inlet, magnetic stirring bar, and reflux condenser was cooled to 0°C. A BH3·THF solution (10 mmol, 4.0 mL of a 2.5 M) was injected into the reaction flask via a syringe, followed by the slow addition of a solution of ß-nitrostyrene in THF (10 mmol, 1.5 g in 20 mL of THF). After the addition, the ice bath was removed and a catalytic amount (~30 mg) of NaBH4 was added to the stirred reaction mixture by means of a spatula. A moderately exothermic reaction ensued. The reaction was allowed to proceed until the yellow color of the starting material disappeared (1 h). Ice-water (100 mL) was added to reaction mixture that was then acidified with 10% HCl (~20 mL). The mixture was stirred, heated at 60-65°C for 2 h, and then cooled to room temperature. The acidic layer was washed with ether (3x50 mL) and then the hydroxylamine liberated via the addition of sodium hydroxide (aqueous). Solid NaCl was added and the product extracted into ether. The combined ethereal extracts were dried over anhydrous MgSO4 and the solvent removed under reduced pressure to yield 1.1 g (80%) of N-hydroxy-2-phenylethanamine: mp 85-86°C (lit.5 83-84°C,6b 84-85°C); melting point of the oxalate salt 171-173°C (lit.6b 170-175°C dec).
N-Cyclohexylhydroxylamine
1-Nitro-1-cyclohexene (2 mmol, 0.254 g) dissolved in 6 mL of THF and BH3·THF (2 mmol, 0.8 mL of a 2,5 M solution) were mixed, and a catalytic amount of NaBH4 was added as described in the general procedure to yield (30 min) 0.216 g (85%) of N-cyclohexylhydroxylamine: mp 137-138 °C (lit.4b 138-139°C); melting point of the hydrochloride 137-142°C dec (lit.12 142°C).
N-Hydroxyamphetamine
ß-Methyl-ß-nitrostyrene (4 mmol, 0.652 g) dissolved in 14 mL of THF and BH3·THF (4 mmol, 1.6 mL of a 2.5 M solution) were mixed, and a catalytic amount of NaBH4 was added to yield (2 h) 0.447 g (74%) of N-hydroxyamphetamine: mp 61-62°C (lit.6b 60.5-62°C); melting point of the oxalate salt 172-173°C (lit.6b 167-169°C.
N-Hydroxy-p-bromoamphetamine
p-Bromo-ß-methyl-ß-nitrostyrene (3 mmol, 0.726 g in 10 mL of THF) and BH3·THF (3 mmol, 1.2 mL of a 2.5 M solution) were mixed, and a catalytic amount of NaBH4 was added to yield (1 h) 0.540 g (78%) of N-hydroxy-p-bromoamphetamine: mp 60-61°C; melting point of the oxalate 169-174°C dec.
N-Hydroxy-p-nitroamphetamine
A mixture of p-nitro-ß-nitro-ß-methylstyrene (5 mmol, 1.04 g) in 25 mL of THF and BH3·THF (5 mmol, 2.0 mL of a 2.5 M solution) was treated with catalytic amount of NaBH4 to yield (1.5 h) 0.750 g (77%) of N-hydroxy-p-nitroamphetamine: mp 71-72°C; melting point of the oxalate 185-186°C.
N-Hydroxy-p-(diethylamino)amphetamine
A mixture of p-(diethylamino)-ß-methyl-ß-nitrostyrene (1 mmol, 0.234 g) in 6 mL of THF and BH3·THF (1 mmol, 0.4 mL of a 2.5 M solution) was treated with a catalytic amount of NaBH4 to yield (3 h) 0.19g (85%) of N-hydroxy-p-(diethylamino)amphetamine: melting point of the oxalate 125-126°C.
N-Hydroxy-2,3-dimethoxyamphetamine
A mixture of 2,3-dimethoxy-ß-nitrostyrene (8 mmol, 1,784 g) in 25 mL of THF and BH3·THF (8 mmol, 3.2 mL of a 2.5 M solution) was treated with a catalytic amount of NaBH4 to yield (1 h) 1.182 g (70%) of N-hydroxy-2,3-dimethoxyamphetamine: mp 66-67°C; oxalate mp 132-133°C.
N-Hydroxyanthracene-9-ethanamine
A mixture of 9-(ω-nitrovinyl)anthracene (10 mmol, 2.5 g) in 20 mL of THF and BH3·THF (10 mmol, 4.0 mL of a 2.5 M solution) was treated with a catalytic amount of NaBH4 (1 h). When an ether extraction of the acidic aqueous layer was attempted, a pale yellow precipitate appeared that was filtered, washed with H2O and dried to yield 1.532 g (56%) of the hydrochloride of N-hydroxyanthracene-9-ethanamine: mp 178-184°C dec. The aqueous filtrate was neutralized with aqueous NaOH and a precipitate was obtained which was filtered, washed with H2O, and dried to yield an additional 0.43 g (18%) of N-hydroxyanthracene-9-ethanamine: mp 126-128°C dec.
{kind=link}