Introduction
A very common way of turning a propenylbenzene into a phenyl-2-propanone (as exemplified here with the conversion of isosafrole to 3,4-MethyleneDioxyPhenyl-2-Propanone,
MDP2P) is to oxidize the alkene (propenylbenzene) using a peracid. A peracid is a carboxylic acid which has been reacted with hydrogen peroxide to form a
highly reactive peracid, which is able to transfer one of its oxygen atoms to the alkene, forming an epoxide (a three-membered ring containing two carbon
atoms and one oxygen8. The peracid is formed like this, exemplified by the conversion of formic acid to performic acid:
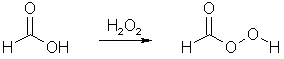
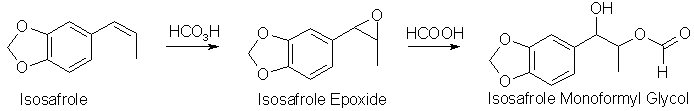
If performic acid is added to isosafrole (or any other propenylbenzene), the first step that happens is that the epoxide is formed. The three-membered
epoxide is a strained molecule, and easily opens up in the presence of a strong acid, and formic acid is a strong enough acid to do that. Formic acid is
always present in the solution, often as a solvent, or in any case it is formed from performic acid when it has transferred its extra oxygen atom to the
alkene. The result is an ester between the isosafrole glycol, 1-(3,4-methylenedioxyphenyl)- 1,2-propanediol, and formic acid, here called isosafrole
monoformyl glycol. This is the main reaction product gotten in this reaction. There are other oxidation products present, like residual epoxide, and some
of the glycol without the ester group present, as well as some impurities gotten from over-oxidation of the isosafrole.
To convert the isosafrole glycol ester to MDP2P, the reaction mixture is heated for a few hours with a dilute (15% w/w) aqueous sulfuric acid (H2SO4) solution, where the ester
first is hydrolysed to isosafrole glycol, which under the influence of the strong acid undergoes dehydration (water elimination) and rearrangement to form MDP2P in a reaction called
the Pinacol RearrangementThe Pinacol Rearrangement
.
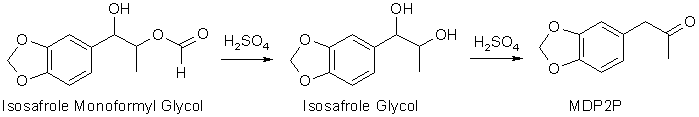
Almost any peracid can be used to oxidize alkenes, one of the most often
used on small scales in professional laboratories is m-chloroperbenzoic acid,
which is relatively stable in its pure state, or magnesium monoperoxy- phtalate.
The advantage is that they do not have to be prepared immediately before use,
but the drawback is their high molecular weight (very much of the peracid
would be needed on a large scale) and that they are much more expensive than
performic acid. One good alternative to performic acid is peracetic acid,
which is prepared from the commonly available acetic acid. Both the concentrated
form (Glacial acetic acid, GAA) and the dilute 90% acetic acid can be used.
As acetic acid is not as strong an acid as formic acid, a strong acid catalyst
is needed to generate the peracetic acid, usually a few ml of sulfuric acid
is added to a mixture of GAA and H2O2, and the well-mixed
solution is allowed to stand overnight9. Often
it is desirable to neutralize the strong sulfurc acid before use, which is
effected by the addition of two equivalents of sodium acetate, which then
results in the formation of neutral sodium sulfate and acetic acid, alternatively
the peracetic acid can be freed of the remaining H2SO4
by flash vacuum distillation10 (at low tempperature
to avoid the risk of explosions). Anhydrous solutions of peracetic acid can
also be made by subjecting the mixture to azeotropic vacuum distillation to
remove the water10.
If the reaction mixture is not as acid as the one in the regular performic
reaction (for example if the reaction is buffered, or a weaker peracid is
used), it is possible that the intermediate epoxide is not ring-opened to
the glycol ester, but that is no problem, as sulfuric acid can open the epoxide
too and convert it to the desired MDP2P. If pure epoxide is isolated, it can
be isomerized to MDP2P, for example through reflux with lithium iodide in
ethyl acetate, but it cannot convert the glycol or the glycol ester to MDP2P.
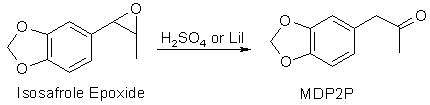
It can be of advantage to buffer the performic acid reaction mixture with a buffering agent (such as sodium bicarbonate), as when the pH is higher, the
reactivity of the peracid is lower, thereby lowering side-reactions due to overoxidation of the product (piperonylic acid and piperonylacetic acid may be
formed, along with the respective aldehydes). It is also important to control the temperature of the reaction, as high temperatures also favors side-reactions.
The yields from reactions with those precautions taken is generally higher (by about 20-25%) than without.
Acidity of carboxylic/percarboxylic acids
- Formic Acid - pKa 3.6
- Performic Acid - pKa 7.1
- Acetic Acid - pKa 4.8
- Peracetic Acid - pKa 8.2
Adding a weak base such as sodium carbonate or sodium bicarbonate (which upon contact with formic acid is converted to carbon dioxide and sodium formate)
does not affect the peracid, as it is a much weaker acid than its corresponding carboxylic acid. The buffer only affects the acidity of the solution caused
by the carboxylic acid solvent, which also is our goal.
The procedures in this document all pertains to the oxidation of isosafrole to MDP2P, but they can be used on any propenylbenzene (such as anethole,
isoapiole, isodillapiole and isomyristicin), except asarone. Asarone is a very acid-sensitive molecule, so it is clear that it won't work in a regular
performic acid oxidation. It has not yet been proven if asarone cannot be used in a peracetic acid oxidation or a buffered performic. It would be
interesting to hear about what would happen in a buffered peracetic acid oxidation.
Performic Acid
Standard Performic acid Oxidation1,2,3
This has been the most common procedure for the peracid oxidation used, because it was the one appearing in
Pihkal1, Total Synthesis II2
and other popular books on MDMA manufacture, such as The Complete Book of Ecstasy. It was pioneered by
the japanese chemists Fugisawa and Deguchi3 in the 1950's. The yield is generally 50-60% of theory. The acetone in the reaction
mixture serves as a co-solvent, making it easier for the isosafrole to mix with the aqueous performic acid mixture. There is a drawback to this, and that is
the possibility of the acetone reacting with the peracid forming acetone peroxide, a very unstable explosive. It is also very tedious to remove all the
excess formic acid by vacuum distillation (it cannot be distilled at ordinary pressure, or there is a risk of decomposition).
Experimental:
To a well stirred solution of 34 g of 30% hydrogen peroxide in 150 g 80%
formic acid there was added, dropwise, a solution of 32.4 g isosafrole in
120 mL acetone at a rate that kept the reaction mixture from exceeding 40°C.
This required a bit over 1 h, and external cooling was used as necessary.
Stirring was continued for 16 h, and care was taken that the slow exothermic
reaction did not cause excess heating. An external bath with running water
worked well. During this time the solution progressed from an orange color
to a deep red. All volatile components were removed under vacuum which yielded
some 60 g of a very deep red residue. This was dissolved in 60 mL of MeOH,
treated with 360 mL of 15% H2SO4, and heated for 3
h on the steam bath. After cooling, the reaction mixture was extracted with
3x75 mL Et2O, the pooled extracts washed first with H2O
and then with dilute NaOH, and the solvent removed under vacuum The residue
was distilled (at 2.0 mmHg/108-112°C, or at about 160°C at the water
pump) to provide 20.6 g of 3,4-methylenedioxyphenylacetone as a pale yellow
oil. The oxime (from hydroxylamine) had a mp of 85-88°C. The semicarbazone
had a mp of 162-163°C.
Performic acid Oxidation (using improved workup)
This procedure circumvents the problem of having to strip off a whole lot of formic acid under vacuum (which takes a long time and corrodes vacuum
pumps) by flooding the reaction mixture with water and extracting the product of the first reaction with an organic solvent. Other people, most notably
Eleusis, has promoted the practice of flooding the reaction mixture with a sodium
hydroxide solution instead to neutralize the formic acid, but this has the disadvantage that it will hydrolyze the glycol ester to the glycol before
the reaction with sulfuric acid, which in many cases will decrease the yield of the reaction. The yield of the reaction described below is a little low,
but the reaction usually gives 50-60% of theory. This method does not scale up so well, or there will be a huge amount of aqueous solution to extract.
Experimental:
A solution of 65 grams of isosafrole in 100ml of acetone was added in one
portion to a stirred solution of 60ml of 35% H2O2
in 300g of 85% formic acid in a 1000 ml beaker situated in an ice-bath (both
solutions was pre-cooled to -20°C in the freezer). Some ice was added to
the reaction mixture to prevent the temperature from rising above 40°C,
(three ice-cubes was needed until the temp stabilized). The mixture was
allowed to stir for 16 hours, whereupon it was poured into 1500ml of cold
water. The cloudy solution was extracted with 3x100ml CH2Cl2,
and the pooled extracts was freed from solvent by distillation.
The dark red residue was taken up in 120 ml of methanol, and added to 700ml
of 15% H2SO4 (w/v), and the solution was slowly refluxed
for three hours. The reaction mixture was cooled and extracted with 3x75ml
CH2Cl2. The extracts were washed with 250ml of water
and 250 ml of 5% NaOH solution. The organic phase was dried over MgSO4,
filtered with suction, the filter cake washed with a little CH2Cl2,
and the solvent was removed by distillation. The black residue was distilled
with 15 mmHg aspirator vacuum, to give ~30 ml of a yellow- brown oil (bp
115-170°C), which was redistilled to give 31g (44%) of an intensely yellow
oil (bp 140-150°C) which gave a single spot with TLC.
Buffered Performic acid Oxidation4
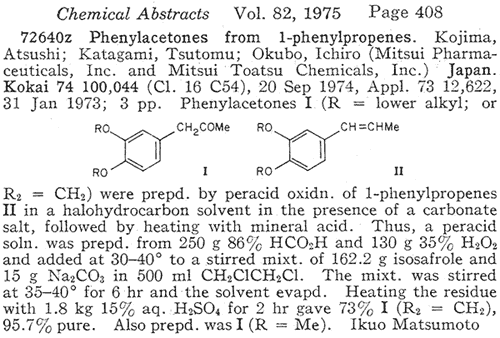
In an article in Chemical Abstracts from 19754
there was a synopsis of an isosafrole oxidation which employed the addition
of performic acid to a suspension of sodium carbonate in a solution of isosafrole
in dichloroethane and reacting for 5h, in which the yield was given as being
73% of theory. A little experimentation by The Hive Collective (most notably
Ritter, Gyro Gearloose and Baalchemist) showed that using twice the molar
amount of sodium bicarbonate (NaHCO3) instead of the sodium carbonate
(Na2CO3) as well as using dichloromethane as the solvent
was preferable, both yield-wise and from a practical viewpoint. The temperature
of the reaction micture should not exceed 40°C, and by using the more common
dichloromethane as solvent, this is taken care about automatically, as that
is its boiling point. It is very important to ensure good stirring in this
reaction, so that the organic and aqueous layers are mixed intimately during
the whole reaction. Another good thing with this variation is that much less
solvents and performic acid is needed. The yield from this reaction is also
one of the highest recorded, ranging between 65-75% of theory.
Experimental:
Isosafrole (230g, 1.42 mol) was dissolved in 550ml dichloromethane in a
2000ml round-bottomed flask (equipped with a reflux condenser and an addition
funnel), and 71g (0.82 mol) NaHCO3 was added to the solution.
Then, a solution of performic acid which was prepared one hour in advance
by mixing 220g (2.25 mol) 35% H2O2 and 290ml (350g,
6.45 mol) 85% HCOOH, was added dropwise to the isosafrole solution during
2h, causing the solution to evolve carbon dioxide and reflux slightly. The
mixture was allowed to stir at room temperature for 16h, and was then poured
into a 2000ml separatory funnel. The bottom organic layer was tapped off
and the top aqueous layer was washed in the funnel with 100ml dichloromethane,
that DCM extract also tapped off and added to the organic layer, which then
was washed with 2x200ml water and 150ml brine (concentrated aqueous sodium
chloride). The light yellow aqueous extracts were backwashed with 75 ml
dichloromethane, and the organic phases were pooled and stripped of solvent
by distillation on a water bath.
The residual orange oil was dissolved in 400ml methanol and lightly boiled
in a 2000ml round-bottomed flask with 1400ml 15% H2SO4
for 2h with good stirring. The solution was allowed to cool, the dark bottom
layer drawn off, and the aqueous layer extracted with 3x200ml diethyl ether.
The pooled organic layers was washed with 3x200ml water, 300ml 5% NaOH and
200ml brine, and dried over MgSO4 and filtered. The ether was
distilled off and the residue vacuum distilled at aspirator vacuum (~15 mmHg) to yield 174g (68%)
of yellow MDP2P (bp 130-150°C).
{kind=link}