Synthesis of 2,5-Dimethoxy-4-methylamphetamine (STP / DOM)IntroductionThe preparation of STP (DOM) in Pihkal suffers from a very bad yield of the intermediate aldehyde and its tedious workup. This synthesis is easier and has much improved yield (88% compared to 25% in Pihkal). 2,5-Dimethoxytoluene1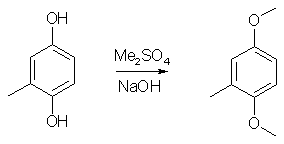
1000ml of water was poured into a wide-mouthed 2000 ml flask, and magnetic stirring was initiated. In sequence, there was added 62 g toluhydroquinone, 160 ml 25% NaOH solution and finally 126 g of dimethyl sulfate. The flask was covered with aluminum foil and stirred for a few days. The brown solution was poured into 2500ml water, and the resulting solution was extracted with 3x100ml of CH2Cl2 and the pooled extracts was stripped of solvent under vacuum. The residue was distilled at about 70°C at 0.5 mmHg to give 49 grams (64%) of 2,5-dimethoxytoluene as a white liquid, which turns orange under several years storage. 2,5-Dimethoxytoluene6A mixture of 12 g of toluhydroquinone, 45 g of potassium carbonate and 45 g of dimethyl sulfate in 300 ml of anhydrous acetone is heated to reflux for 4 days. After cooling, the reaction mixture is filtered and the filtrate is evaporated under vacuum. The residue is taken up in 150 ml of concentrated aqueous ammonia, the mixture is left stirring for 2 hours, diluted with water and extracted with DCM, the organic phase is dried over magnesium sulphate and the solvent is evaporated off under vacuum. The residue is chromatographed on silica gel H, eluting with a heptane/DCM (50:50; v/v) mixture. 12g (55%) of the expected product is obtained. 2,5-Dimethoxytoluene7To a well-stirred solution of NaOH (44 g, 1.1 mole), and Na2S2O4 (8.7 g, 50 mmol) in water (500 mL) under N2, was added toluhydroquinone (62.07g, 0.50 mole). To the homogeneous solution Me2SO4 (99.4 mL, 1.05 mole) was added and the mixture stirred for 0.5 hour. (If the solution did not remain basic, more NaOH was added, and the mixture stirred for 0.5 hour more). The aqueous layer was extracted with ether (2x200 mL), and the ether layer was washed with 1N NaOH (200 mL). The ether layer was dried (MgSO4), filtered, and the solvent was removed. After vacuum distillation, and the solvent was removed. After vacuum distillation, the yield was 54.9g (69%). 2,5-Dimethoxy-4-methylbenzaldehyde2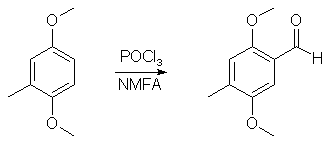
40ml POCl3 and 45ml N-methylformanilide was stirred at room temp for 50 min to give a nice orange solution of the Vilsmeyer complex. 15.2g (0.1 mole) 2,5-dimethoxytoluene was then added in one portion, and the reaction mixture was heated to 70°C, during which time the reaction mixture became dark red. The heat source was then removed, and the exothermic reaction, which evolved large amounts of HCl gas, was allowed to go on until the mixture cooled down and solidified. The flask was then heated on the steam bath for 2 hours, and the warm reaction mixture poured into 2 liters of ice water, and was stirred until all the ice had melted. The ice-cold solution was filtered, and the crude aldehyde was collected as a red solid. This was suspended in 50ml of boiling petroleum ether and was decanted. The procedure was repeated two more times with 50ml of petroleum ether each. This left a small amount of red goo on the bottom of the flask. The solution was allowed to crystallize in the fridge for an hour and was then filtered. The mother liquor was concentrated to 25% of the original volume, again cooled in the fridge and filtered. The orange-brown precipitate was then recrystallized from 50 ml 80% aqueous MeOH to give 14.6g (88%) of 2,5-dimethoxy-4-methylbenzaldehyde as orange crystals. 2,5-dimethoxy-4-methylphenyl-2-nitropropene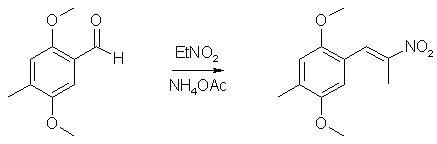
To 50 ml glacial acetic acid in a 100ml flask there was added 10.8 g of 4-methyl-2,5-dimethoxybenzaldehyde, 7.5 g nitroethane, 4 g ammonium acetate and 2 ml of acetic anhydride. The solution was heated on a water bath for 3h, whereupon the solvent was removed under vacuum. The residue was mixed with 100 ml H2O and extracted with 2x50ml CH2Cl2. The organic phase was washed with 50ml 10% NaHSO3 followed by 50ml H2O. The pooled aqueous phases was extracted with 50 ml CH2Cl2, the organic phases dried over MgSO4, and the solvent was evaporated under vacuum. The residue was dissolved in 50ml IPA and was allowed to precipitate in the freezer to give 8.15 g of orange crystals (69%). Alternative method: Use ethylenediamine diacetate in isopropanol as the catalyst instead, giving 80% yield with a lot less tedious workup. 2,5-Dimethoxy-4-methylamphetamine3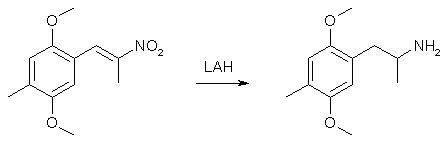
In a two-necked round-bottomed flask, equipped with a reflux condenser and a calcium chloride drying tube, a suspension of 9.5 g LAH in 750 mL well stirred anhydrous Et2O was brought to a boil, and a THF solution of 9.5 g 2,5-dimethoxy-4-methylphenyl-2-nitropropene was added dropwise through an addition funnel. After the addition of the nitrostyrene was complete, the stirred suspension was maintained at reflux for an additional 4 h with a water bath, then allowed to cool to room temperature and the stirring was continued overnight. The excess hydride was destroyed by the addition of 750 mL 8% H2SO4, cautiously, until the hydrogen evolution ceased, then at a speed that allowed the formed solids to disperse. The phases were separated, the aqueous phase washed once with Et2O, treated with 225 g potassium sodium tartrate, and finally made basic (pH > 9) with 5% NaOH. This was extracted with 3x150 mL CH2Cl2, the extracts pooled, and the solvent removed under vacuum. The residue was 9.6 g of a clear oil which spontaneously formed crystals with a mp of 60.5-61°C from hexane. These solids were dissolved in 150 mL anhydrous Et2O, and saturated with anhydrous HCl gas. After standing at room temperature for 2 h, the crystalline 2,5-dimethoxy-4-methylamphetamine hydrochloride (DOM) was removed by filtration, washed with Et2O, and air dried to constant weight. There was obtained 8.25g (84%) of glistening white crystals that had a mp of 190.5-191.5°C. Synthesis of 2,5-Dimethoxy-4-methyl-propenylbenzene4(a) With 1 Equiv of Base.Ethyltriphenylphosphonium bromide (11.10 g, 0.03 mol) in 50 mL of dry THF was stirred under a N2 atmosphere with 3.36 g (0.03 mol) of commerical potassium tert-butoxide. The mixture was stirred for 2 h, resulting in an orangered colored solution. A solution of 2,5-dimethoxy-4-methylbenzaldehyde (5.40 g, 0.03 mol) in 25 mL of dry THF was added dropwise to the reaction mixture. As the addition proceeded, the color changed to bright yellow. The mixture was stirred a t room temperature for 0.5 h and refluxed overnight under an N2 atmosphere. The reaction mixture was filtered, diluted with water, and extracted three times with CHCl3 The combined extracts were washed with brine, dried (MgSO4), filtered, and concentrated. The residue was taken up into a minimum of Et20, and the solution was decanted to remove triphenylphosphine oxide, which separated as a solid. The solid was washed with ether, and the combined ether solutions were concentrated and passed through a short silica gel column eluted with ethyl acetate-hexane (l:l), to remove remaining traces of triphenylphosphine oxide. The required olefin, along with some unreacted aldehyde, was rapidly eluted from the column. Concentration of this material gave a residue, which was taken up into 10 mL of EtOH and treated with 10 mL of saturated NaHSO3 solution. The bisulfite adduct was removed by filtration and was repeatedly washed with ether. The combined filtrate and washings were concentrated to give the required product as a yellow liquid: yield 3.56 g (62%); bp 110-125°C (0.15 mm); GC analysis indicated the product to be 72% Z isomer and 28% E isomer. (b) With 2 Equiv of Base.The Wittig reaction was repeated as above but using 2 equiv of potassium tert-butoxide. Thus, 5.4g (0.03 mol) of the aldehyde and 11.1 g (0.03 mol) of ethyltriphenylphosphonium bromide with 6.72 g (0.06 mol) of potassium tert-butoxide yielded, after distillation, 2.89 g (50%) of the pure olefin, bp 105-110°C (0.05 mm). GC analysis showed it to be 97% E and 3% Z. 4-Methyl-2,5-Dimethoxypropiophenone5To a solution of 15.2g (0.1 mole) of 2,5-dimethoxytoluene and 9.2g (0.1 mole) of propionyl chloride in 125ml carbon disulfide was added portionwise 13.4g (0.1 mole) of AlCl3 at such a rate that the temperature of the reaction mixture remained between 0-10°C. The addition required about 30 minutes. After stirring at room temperature for three hours, the dark green mixture was decomposed by pouring into 80ml of crushed ice and 5ml conc HCl and then filtered to yield 3.1g of a solid, mp 76-77°C. The filtrate was extracted with 2x50ml CHCl3, the extracts combined, dried over Na2SO4 and evaporated in vacuo leaving 15.6g, mp 76-77°C, which was recrystallized from 95% Ethanol to give 8.4g ketone, mp 76-77°C. Concentration of the mother liquor gave a third crop of 5.6g, mp 76-77°C, together adding up to a total yield of 17.1g (82%). References [1] Shulgin & Shulgin, Pihkal #23, p 511
|